
No longer weary, stale, flat, and unprofitable, protein surfaces can be altered to improve drug targeting and screening, enhancing the “druggability” of protein-protein interactions
In protein-protein interactions (PPIs), the participating proteins may have more activity, or less, depending on whether they have undergone a modification of some kind. The modification may be one that has been introduced deliberately, in which case its purpose is likely therapeutic. In fact, most protein-based pharmaceuticals derive their therapeutic effect from some sort of modification.
Assessing the effects of deliberate modifications is crucial to the design and selection of lead molecules for future therapeutics. Major research and commercial efforts are focused on evaluating structural modifications that activate, inhibit, or stabilize proteins, and proteins of all kinds are of interest—enzymatic kinases, proteases, transcription factors, and so on.
As you might expect, the methods used to introduce and evaluate protein modifications are varied. They are also becoming more precise, turning PPIs into frameworks for pharmaceutical analysis and the realization of efficacious therapeutics. The methods highlighted in this article demonstrate that investigators are working at multiple levels. Some investigators are developing chemical “warheads” to target varied amino acid residues; some are pairing proteins with protein fragments via molecular “tethers”; and some are integrating biophysical and functional approaches in their work. In every instance, the goal is to find “druggable” features in seemingly unpromising molecular landscapes.
A target-rich PPI environment
Maurizio Pellecchia, PhD, professor of biomedical sciences at the UC Riverside and director of UC Riverside’s Center for Molecular and Translational Medicine, is developing agents that target amino acids besides cysteine (Cys)—lysine (Lys), tyrosine (Tyr), and histidine (His)—to make more potent irreversible PPI antagonists.
“Our goal,” he says, “is to obtain well-validated pharmacological tools to use as stepping stones for continued drug development via biotechnology or pharmaceutical companies.” Starting with weakly interacting initial agents, Pellecchia and colleagues use biophysics-based screening techniques (such as nuclear magnetic resonance spectroscopy, isothermal titration calorimetry, and thermal shift analysis) to iteratively optimize the targets to form irreversible PPI antagonists.
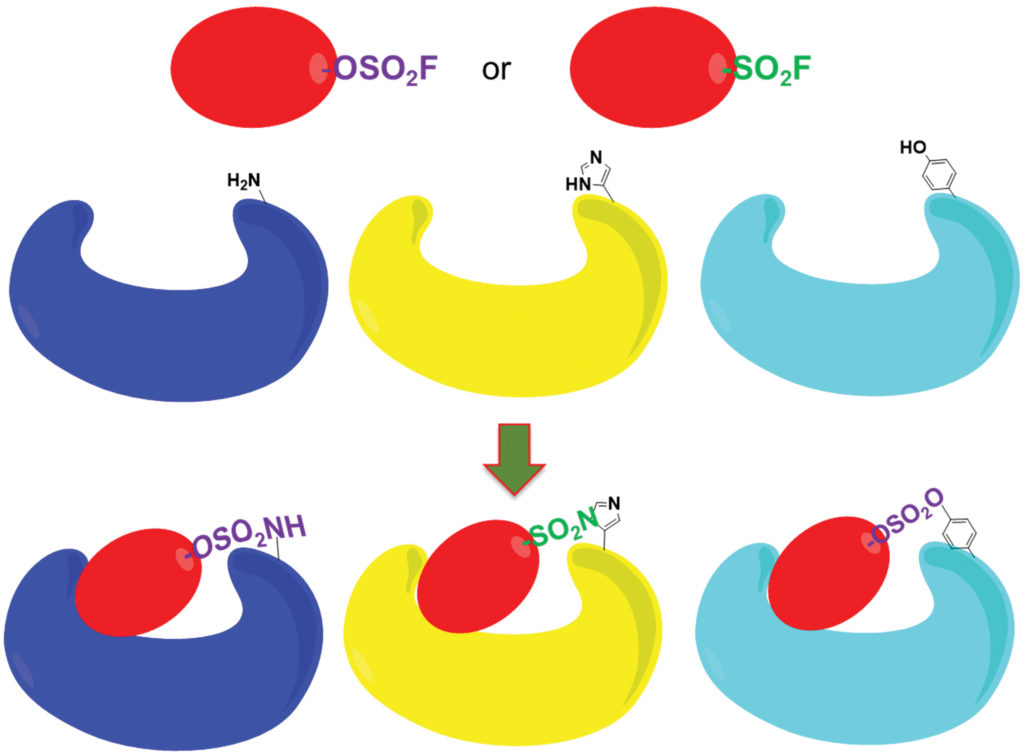
To demonstrate his approach, Pellecchia is applying it to molecules such as X-linked inhibitor of apoptosis protein (XIAP). Progress depends on finding electrophiles that form covalent adducts with proteins. (These electrophiles form covalent bonds with the proteins’ exposed amino acids.) Pellecchia and colleagues are extending existing techniques such as the targeting of Cys residues by the mild electrophile acrylamide to create irreversible enzyme inhibitors. To target exposed Lys, Tyr, and His residues, the investigators are using electrophiles such as aryl-fluorosulfates (-OSO2F) and sulfonyl fluorides (-SO2F).
“Unlike Cys,” Pellecchia says of Lys, Tyr, and His, “these additional amino acids are more often found at the binding interfaces of PPI proteins. If successful, this approach could significantly expand the space for the druggable proteome.”
His team recently published a paper describing the approach for targeting Lys in the apoptosis regulator XIAP. Another manuscript describes Cys covalent agents targeting a Bcl-2 family of apoptosis regulators. He is currently working on developing similar agents targeting Lys, His, or Tyr of other oncology targets such EphA2 and other Bcl-2 proteins.
At the start of one’s tether
Binding a target protein with a covalently bound protein fragment is an effective way to create an antagonist of the natural protein. But as Daniel A. Erlanson, PhD, vice president of chemistry at Frontier Medicines suggests, making the target “druggable” requires not just knowing “whether a compound is bound to a protein” but “where on the protein it’s bound.” For Erlanson, this means performing fragment-based drug discovery (FBDD), which uses techniques such as mass spectrometry to detect associations in small molecular fragments, that is, fragments weighing less than 250 Da and including fewer than 18 nonhydrogen atoms.
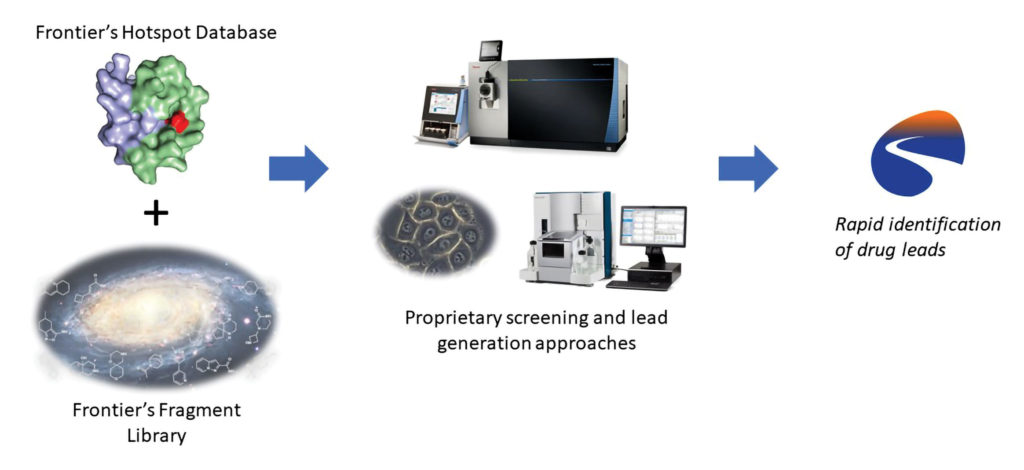
“The strength of FBDD approaches is that they can give you a starting point when conventional screens wouldn’t turn up anything,” Erlanson states. “You take that starting point and build it up or elaborate it into a bigger molecule.” Small fragments, he argues, mean fewer choices to consider and more efficient searches of chemical space.
Erlanson is reluctant to discuss his lead molecules at Frontiers Medicines, but he is willing to offer a suggestion: “Once you’ve built up fragments into molecules approaching drug-like affinity, those molecules are further optimized into lead drug candidates.”
He notes that recent investigations used FBDD to create a druggable target of KRAS. One of these investigations involved a collaboration between Carmot Therapeutics (Erlanson’s former company) and Amgen. Other investigations have been carried out independently by Kevan M. Shokat, PhD, and colleagues at the University of California, San Francisco. (Shokat’s group uses a technique known as tethering, which Erlanson co-developed.) According to Erlanson, “Combining information from these efforts led to the first clinical-stage KRAS inhibitor, AMG 510.”
BAX to BAX studies of apoptosis inhibition
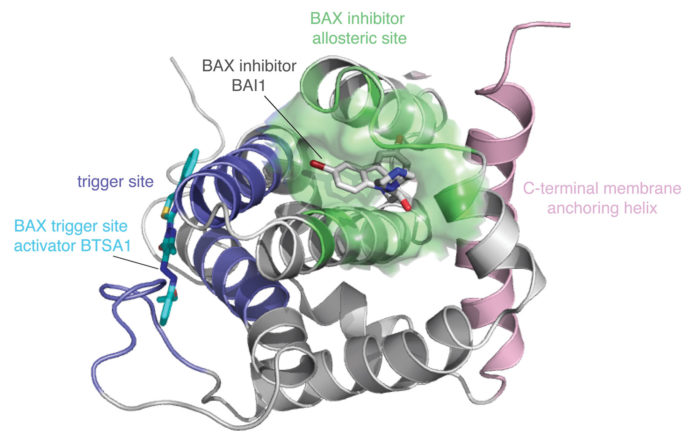
At the Albert Einstein College of Medicine, a research team led by professor of biochemistry Evripidis Gavathiotis, PhD, has been examining the PPIs of the Bcl-2 family of proteins. These PPIs mediate apoptosis and autophagy. Hence, they influence life-and-death decisions in healthy and malignant cells. By discovering the mechanistic details behind these PPIs, the research team hopes to identify drug targets that could inform the development of activators and inhibitors of Bcl-2 proteins.
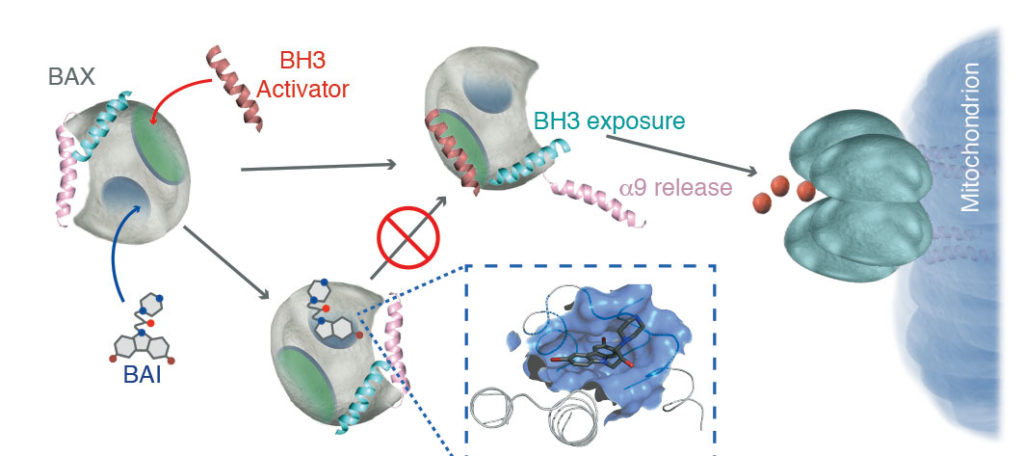
Gavathiotis and colleagues have been particularly focused on Bcl-2-associated X protein (BAX), a molecule known to stimulate apoptosis. To identify potential BAX inhibitors, the investigators evaluated biophysical interactions between molecular fragments of BAX-associated proteins.
The team uses NMR and other biophysical techniques to thoroughly characterize interactions. “We place a significant amount of effort on the structural characteristics of new mechanisms for small molecules,” Gavathiotis points out.
The inhibitors his team has discovered include small-molecule inhibitors that bind directly to a previously unrecognized BAX pocket and act allosterically. These inhibitors, which Gavathiotis and colleagues described last year in Nature Chemical Biology, have now been investigated in animal models. The results, which were presented in a recent paper in Nature Cancer, indicate that one of the team’s BAX inhibitors protects against chemotherapy-induced toxicity—specifically, doxorubicin-induced cardiomyopathy. “Cardioprotection,” the authors of the paper noted, “does not compromise the efficacy of doxorubicin in reducing leukemia or breast cancer burden in vivo, primarily due to increased priming of mitochondrial death mechanisms and higher BAX levels in cancer cells.”
To summarize, the Gavathiotis team’s work on the BAX inhibitor progressed from biophysical characterization to functional evaluation. The team began by detailing how the BAX inhibitor’s binding causes conformational changes in BAX that prevent mitochondrial translocation and oligomerization. Then the team evaluated the BAX inhibitor’s potential for in vivo activity. While relying on animal models of leukemia and breast cancer, the investigators used commercial caspase to assess viability, apoptosis, and mitochondrial-mediated necrosis.
A complementation approach to cell-based assays
To aid in the cellular analysis and screening of novel druggable targets, companies like Eurofins DiscoverX are producing cellular assays that themselves use PPIs to advance the screening of lead molecules. Jane Lamerdin, PhD, director of R&D at Eurofins DiscoverX, aids investigators through the production of cellular assays with their novel ligand-induced PPI systems. She says, “We usually engineer our assays so that they capture receptor activation by looking at recruitment of a particular scaffolding or signaling protein.”
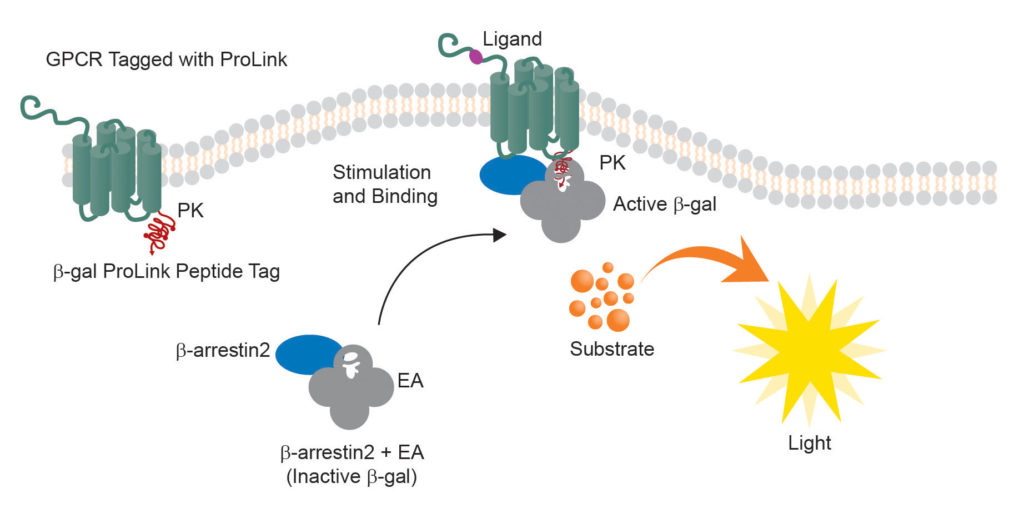
The cell lines produced by Eurofins DiscoverX use a split β-galactosidase reporter, with each piece fused to an interacting protein in a signaling cascade. “We co-express those two components in the cell of interest,” she explains. “When there is no ligand present, there may be some background signal due to the overexpression of the two components, but it is very low. Then, when you stimulate the cells with the appropriate ligand, you see a gain of signal.”
The β-galactosidase activates its substrate only after the two fragments are rejoined through an activation event, such as a ligand-mediated receptor phosphorylation event that recruits an adaptor protein. DiscoverX has been developing these assays for years, meeting demands cited by individual customers or identified through market research.
Historically, these assays have been developed using heterologous constructs, says Lamerdin, “with expression driven by a cytomegalovirus promoter.” However, recent advances in genetic engineering techniques have allowed the company to produce its first CRISPR-Cas9-modified cell lines, using the same reporter technology, which allows evaluation of physiologically relevant levels of the protein of interest, which is critical for evaluating efficacy of certain therapeutics, such as those used to hijack endogenous E3 ligases to degrade oncoproteins or protein aggregates, such as tau.
In the company’s proof-of-concept experiments, “the endogenous protein of interest (in our case BRD4) is tagged with the small β-galactosidase fragment using CRISPR-Cas9,” she says. “We treat the cells with a bifunctional molecule that targets BRD4 on one end and binds to a specific E3 ligase in the cells on the other end to induce degradation of BRD4.”