Super-resolution microscopy provides real-time picture of bacteria degrading biomass with enzyme complexes
To the untrained eye, they look like blobs blotching the otherwise smooth surface of rod-like bacteria. But if you ask a microbiologist about “cellulosomes,” they will likely tell you that those blobs are actually sophisticated cellulolytic machines.
“Only about 10 to 20 microbes have been so far identified with cellulosomes and characterized, so it is a really rare biomass deconstruction mechanism in nature,” said Yannick Bomble, a National Laboratory of the Rockies (NLR) group manager and senior bioenergy researcher who has studied these enzyme structures for years. “C. thermocellum is one of them, and it is the best biomass degrader that has been identified so far. There’s nothing else that comes close.”
Cellulosomes are big protein complexes packed with dozens of unique enzymes. For years, researchers have known that they are core to C. thermocellum’s exceptional ability to degrade raw biomass. But researchers have lacked a functional picture of how bacteria produce and use cellulosomes—a missing clue to optimize C. thermocellum for turning biomass into chemicals and fuels.
Now, in a paper published in Life Science Alliance, NLR researchers and collaborators used super-resolution imaging and machine-learning-based analysis to quantify the location and movement of cellulosomes on C. thermocellum during biomass deconstruction. The team was surprised not only by the abundance of cellulosomes on each bacterium but also by evidence suggesting that the cells dynamically redistribute them to increase their interaction with biomass.
The team’s machine-learning-guided microscopy techniques represent an advantageous approach for further research—unlocking more insights for someday harnessing C. thermocellum in next-generation consolidated bioprocessing systems.
Large-scale image analysis reveals how C. thermocellum reorganizes cellulosomes
Conventional microscopy is labor intensive, and, as senior NLR researcher John Yarbrough is well aware, it only provides a limited snapshot into the life cycle of bacteria.
“Many microscopy studies capture only a handful of data points,” explained Yarbrough, who is the first author on the paper. “The key question is whether those data points truly represent the full sample and the broader biological system.”
Aiming for a more comprehensive understanding of C. thermocellum, the team, which also included NLR researchers Neal Hengge, Qi Xu, Samantha Ziegler, Daehwan Chung, and Shu Huang, employed an advanced microscopy and analysis technique.
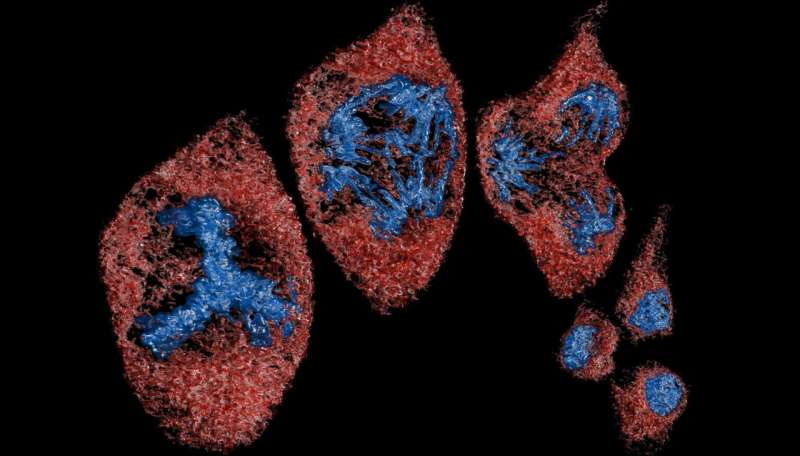
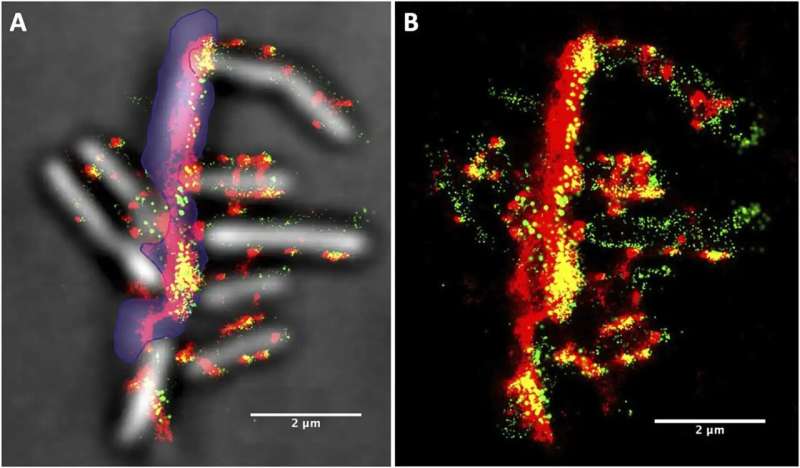
Using a super-resolution microscopy setup—now a well-established capability at NLR that goes beyond traditional optical microscopy to resolve objects barely 30 nanometers apart—they took more than 15,000 images of bacterial samples obtained at different stages of growth. When captured in close succession every 30 milliseconds, each image recorded different signals emitting from the bacteria and cellulosomes, which the team had tagged with fluorescent probes.
Then, the team used an unsupervised machine-learning clustering algorithm, which acts like a digital organizer to autonomously discover hidden patterns, to analyze the location of each signal. The result was a handful of pictures superimposing hundreds of these signals in a single frame—each showing the location and evolution of cellulosomes during different stages of bacterial growth.
“We noticed that there are significantly more cellulosomes on the plant biomass (i.e., lignocellulose) than on the actual bacteria toward the end of the bacteria’s life,” Yarbrough said. “Where the bacteria were binding with the biomass substrate, we measured in some locations over 1,000 cellulosomes tightly bound in a tiny space.”
In other words, the team resolved the spatial distribution of cellulosome clusters and found that they became enriched at the cell surfaces touching the biomass. As biomass deconstruction progressed, the team noticed that the cellulosomes moved—not remaining fixed on the cell surface. They appeared to disengage, and the bacteria became depleted in cellulosomes in later stages of the cellular life cycle.
More than a pretty picture: A statistical look for quantifying improvements
“What’s exciting to me is that, for once, this imaging is quantitative,” Bomble said of the results. “We can build histograms and conduct statistical analysis to actually measure changes in different stages of bacterial growth, see the size of cellulosome clusters, and measure the number of cellulosomes per unit area.”
For a bacterium like C. thermocellum, such quantitative data provide a basis for someday further engineering them. In the context of industry, the study provides ambition for “consolidated bioprocessing,” where bio-based chemicals and fuels are made in a single pot—rather than two or three using conventional methods.
“The traditional way of achieving biomass deconstruction and its upgrading is pretreatment, enzymatic hydrolysis, and conversion of those sugars into products,” Bomble explained. “What’s appealing about consolidated bioprocessing is that there is no biomass pretreatment or added enzymes, which are significant expenses in cellulosic biofuels, so consolidated bioprocessing could be a lot cheaper.”
Through the Center for Bioenergy Innovation, NLR and other national laboratories are connecting the dots between fundamental microbiology traits and improvements needed for consolidated bioprocessing to work. Thanks to the tools developed in this study, the team was able to connect a few more dots for cellulosomes, which Yarbrough said will also facilitate research on other promising bacteria and microbial consortia.
“This technique allowed us to quantify information to compare results as we genetically modify the cellulosomes,” Yarbrough added. “Do we see change during the growth phases? The beauty of this technique is that we can now go back and compare previous data and show in future publications whether the changes we are making to the cellulosomes lead to improvements in performance.”
NLR’s previous research on cellulosomes has already resulted in intellectual property for biotechnology companies, most recently in another advanced method called cell-free biomanufacturing. Bomble and his team have leveraged the components of the cellulosome to array metabolic enzymes for advanced enzyme immobilization—an example of U.S. Department of Energy Office of Science support resulting in breakthroughs for more applied research.
After all, as Bomble knows, cellulosomes are no mere blobs. Cellulosomes are sophisticated, and the microbes that use them hide insights that could improve how chemicals and fuels are made in the United States.
“This microbe is full of surprises and inspiration,” Bomble said. “It is where ideas come from. This is how fundamental science is now making its way into the more applied sciences.”