

With 99% of patients experiencing an adverse event, the FDA is skeptical that the risks associated with Geron’s myelodysplastic syndromes (MDS) medicine imetelstat outweigh the potential benefit of treatment, particularly in patients with lower-risk cases of the blood cancer.
Geron is seeking approval of imetelstat for transfusion-dependent anemia in adult patients with low- to intermediate-risk MDS who have failed to respond to, have lost response to or are ineligible for erythropoiesis-stimulating agents.
The FDA has called on an advisory committee to answer a few key questions about the data submitted to support the approval application. Specifically, the Oncologic Drugs Advisory Committee will be tasked with discussing: the efficacy of imetelstat for the proposed patient population based on the results of the phase 3 MDS3001 trial considering the safety profile and whether the benefits of the drug outweigh the risks.
Imetelstat is a telomerase inhibitor that is designed to bind to telomerase to inhibit its activity, killing the cancerous stem and progenitor cells in the bone marrow that cause MDS.
In the phase 3 MDS3001 study, imetelstat was tested in 118 patients, while 59 received placebo. The therapy was linked with eight-week red blood cell transfusion independence (RBC-TI), which was the main goal of the trial. The trial showed that 40% of patients on the study drug met the eight-week RBC-TI threshold compared to 15% on placebo. At 24 weeks, the difference was 28% for imetelstat and 3% for placebo. An earlier phase 2 study had similar rates of efficacy.
In briefing documents issued ahead of the Thursday advisory committee meeting, the FDA expressed concerns on whether the magnitude and durability of RBC-TI outweighs the risks of treatment for patients with lower-risk MDS. The agency also flagged that secondary endpoint results were not supportive of a disease-modifying treatment effect, including complete remission and partial remission and overall survival.
Patient-reported outcomes of reductions in fatigue or other anemia-related symptoms were not supportive of a treatment effect, according to the FDA.
The FDA also noted that the trial population may not be representative of the U.S. population that the application would seek to serve, as only 13 patients were enrolled here. Of the eight patients on the study drug, just one met the main goal of the study, meaning RBC-TI rates of just 13% compared to 0% in the placebo group of five patients.
On safety, imetelstat spurred treatment-emergent adverse events in 99% of patients—but the placebo group had a rate of 100% as well. The difference is in the severity of the events, with 107 patients taking Geron’s medicine experiencing a grade 3 or 4 event compared to 28 in the placebo arm.
Of primary concern to the FDA is the rates of neutropenia and thrombocytopenia, low neutrophil count and low platelet count, respectively. The rates of these complications were higher in patients taking the study drug than placebo. Patients on Geron’s drug were also more likely to require myeloid growth factor support or platelet transfusion.
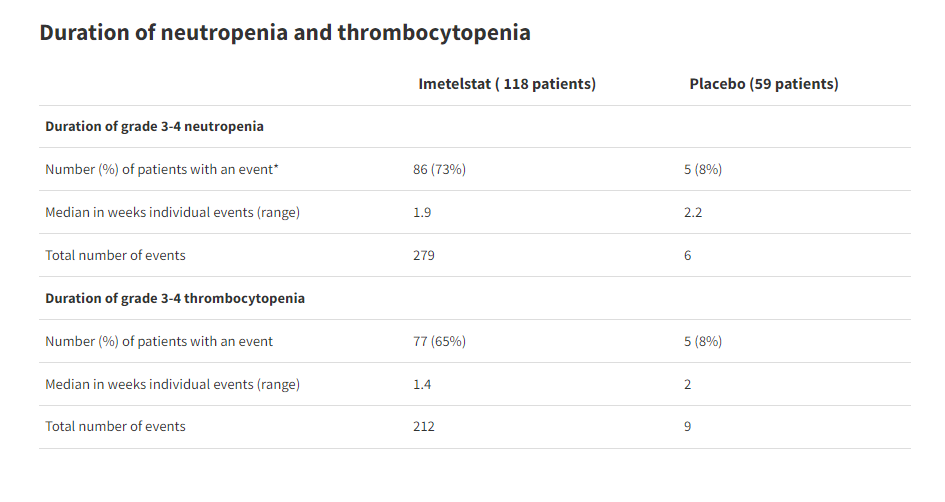
Even with increased supportive care, these patients experienced higher rates of infections and hemorrhagic events than those on placebo, which the FDA said suggests that the neutropenia and thrombocytopenia resulted in clinical consequences.
The serious adverse events led to dose modification, including dose interruption, decrease and discontinuation in the imetelstat arm. The most common events were infections, fatigue, anemia, joint stiffness, muscle aches and hemorrhage.
There was one fatal treatment-emergent event in each arm of the study. In the imetelstat arm, the death was a 72-year-old man with low risk MDS who experienced grade 3 neutropenia and thrombocytopenia on Day 619 of treatment. He received treatment on that day, which the FDA said was a protocol violation. He went into grade 4 sepsis and died of septic shock, respiratory failure, pneumonia and cardiac ischemia 21 days later.
“It is worth noting that cytopenias occurred regardless of whether a patient responded to imetelstat or not. As most patients do not respond to imetelstat, there is a substantial risk of exposing patients to toxicity with no durable RBC-TI,” the FDA wrote.
Two patients also died of complications of infections that happened 30 days after their last dose, which the FDA said indicates that neutropenia and the risk of infection lasts after the end of treatment with imetelstat.
Because of these safety issues, the FDA said that there has not been enough work to determine whether a lower dose might be more appropriate for the lower risk MDS population.
In summary, the FDA said: “Overall, treatment with imetelstat is associated with risks that might be considered substantial, and it is not clear that the risks of treatment with imetelstat are outweighed by the potential benefit for the intended population.”
Geron’s response
In its briefing documents, Geron defended imetelstat’s benefit-risk ratio as “favorable” for the proposed patient population. The company said the risks of neutropenia and thrombocytopenia—which include severe bleeding and infection—are manageable and comparable to placebo.
While there was indeed a higher rate of neutropenia and thrombocytopenia in patients taking imetelstat, Geron noted that this did not translate into higher incidences of the more severe consequences. That’s because patients received the study med for a short period of time, the company said.
“Regarding the safety profile, the known and well characterized risks of grade 3 and 4 neutropenia and thrombocytopenia with imetelstat treatment are manageable by hematologists and health care providers who take care of MDS patients,” Geron said.
The company noted that standard of care treatments for the condition also have high rates of these complications.
When neutropenia and thrombocytopenia were excluded, the rate of grade 3 and 4 serious adverse events “decreased significantly” to 54% for imetelstat and 39% for placebo—however, the FDA disagreed that this was an acceptable rate.
“The clinical studies demonstrated substantial evidence of effectiveness, and the safety profile is well characterized and manageable,” Geron wrote.
Given the unmet need and limitation of approved therapies to very specific subgroups, Geron believes that imetelstat could provide a meaningful treatment option. The biotech also said that the dose is supported by the phase 2 and 3 data.