
Cancer drugs are designed to shut tumors down. But sometimes, in the very act of attacking a tumor, treatment can also help a small fraction of cancer cells become harder to kill. A new study from researchers at the Institute for Systems Biology (ISB) shows that cancer cells may begin escaping therapy much earlier—as the therapy itself triggers a stress response that drives some cancer cells into a temporary drug-tolerant state.
Published in Nature Communications, the study shows that melanoma cells exposed to BRAF-targeted therapy do not simply wait for resistance mutations to emerge.
Instead, they launch an early, coordinated survival program that pushes them into a temporary drug-tolerant state, allowing them to endure treatment long before permanent genetic resistance takes hold.
Using high-resolution, time-series multi-omics and computational modeling, the researchers reconstructed what they describe as a “molecular movie” of this transition—capturing the earliest events that occur within hours to days after treatment begins.
Rather than comparing cells only before treatment and after resistance had already emerged, the team tracked the escape process in real time, revealing that it follows an ordered sequence of events rather than a random drift into resistance.
“We tend to think of drug resistance as something that happens later, after tumors evolve new mutations,” said Wei Wei, Ph.D., co-senior author of the study and associate professor at ISB.
“What we’re seeing here is that the escape process begins almost immediately. Cells actively reprogram themselves to survive the initial shock of therapy.”
A rapid identity shift—not just genetic resistance
The study focused on melanoma driven by mutations in the BRAF gene, a common target of precision therapies. While these drugs can produce strong initial responses, in many patients, the tumors eventually find a way back.
The researchers found that, in response to treatment, melanoma cells undergo a reversible shift away from their original, drug-sensitive identity into a more primitive, therapy-tolerant state. This transition is not random. It unfolds through two sequential “transcriptional waves” that progressively reorganize gene activity and cellular identity.
Even more striking, when the drug is removed, the cells do not simply retrace their steps. They return by a different route, retaining a form of “molecular memory” of prior treatment.
“This tells us that resistance isn’t just about which mutations a tumor has,” said ISB President and Professor Jim Heath, Ph.D., co-senior author of the study. “It’s also about the cell states that treatment itself pushes cancer cells into—and how those states shape future behavior.”
An early molecular trigger: Stress, NF-κB, and chromatin remodeling
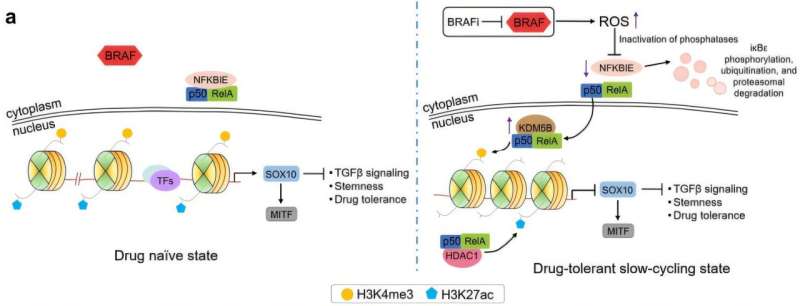
At the center of this adaptive response is NF-κB, a well-known regulator of cellular stress and survival.
The study shows that NF-κB is not just a general marker of cellular distress. It acts as an early trigger that converts the shock of targeted therapy into a survival program.
Specifically, targeted therapy disrupts antioxidant defenses and leads to a buildup of reactive oxygen species (ROS). This oxidative stress activates NF-κB, which then drives widespread changes in gene regulation.
Once activated, NF-κB recruits epigenetic enzymes that modify chromatin—the packaging system that determines which parts of DNA are open for reading and which are closed off.
In effect, the stress response begins to rewrite which genetic instructions the cell can access. One key target is SOX10, a transcription factor essential for maintaining the melanocytic state. As those identity genes are shut down, melanoma cells shift into a drug-tolerant condition that allows them to persist under therapy.
Toward more durable cancer treatments
While the findings are preclinical, they point to a new therapeutic strategy: preventing cancer cells from entering this escape state in the first place.
Rather than waiting for resistance to emerge, the researchers suggest that combining targeted therapies with drugs that disrupt the epigenetic programs downstream of this stress response could help cut off the escape route at its earliest, still-reversible stage.
The team also found evidence that similar stress-driven pathways operate in other cancers, including lung and colon cancer, suggesting that this may represent a broader mechanism of therapy resistance.
More broadly, the work reframes cancer resistance not simply as a genetic problem, but as a dynamic cell-state problem—one in which treatment itself can create the stress conditions that help some tumor cells survive unless that early escape program is blocked.
“Resistance may begin not only when cancer cells acquire new mutations, but when treatment itself pushes surviving cells into a stronger, more evasive state,” Wei said.
“If we can intervene early—at the level of cell-state transitions—we may be able to extend the effectiveness of targeted therapies across multiple cancer types.”